Consequence calling
The BCFtools/csq command is a very fast program for haplotype-aware consequence calling which can take into account known phase. It avoids the common pitfall of existing predictors which analyze variants as isolated events and correctly predicts consequences for adjacent variants which alter the same codon or frame-shifting indels followed by a frame-restoring indels.
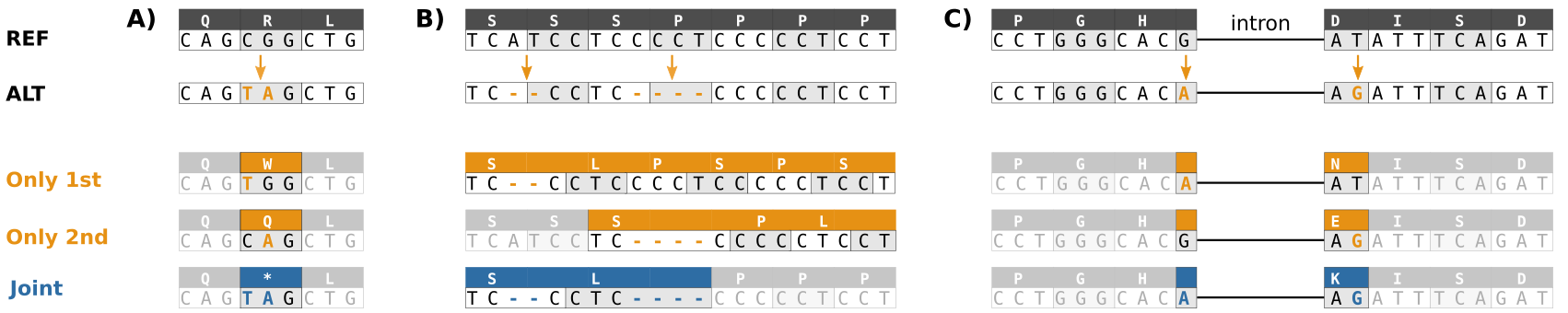
Three types of compound variants that lead to incorrect consequence prediction when handled in a localized manner each separately rather than jointly.
A) Multiple SNVs in the same
codon result in a TAG stop codon rather than an amino acid change. B) A deletion locally predicted as frame-shifting is followed by a frame-restoring variant. Two amino acids are deleted
and one changed, the functional consequence on protein function is likely much less severe. C) Two SNVs separated by an intron occur within the same codon in the spliced transcript.
Unchanged areas are shaded for readability. All three examples were encountered in real data.
The program requires on input a VCF/BCF file, the reference genome in fasta format and genomic features in the GFF3 format downloadable from the Ensembl website, and outputs an annotated VCF/BCF file. Currently, only Ensembl GFF3 files are supported, see for example ftp://ftp.ensembl.org/pub/current_gff3/homo_sapiens.
The typical command looks like this
bcftools csq -f hs37d5.fa -g Homo_sapiens.GRCh37.82.gff3.gz in.vcf -Ob -o out.bcf
The program adds a consequence annotation in a format similar to VEP:
Consequence|gene|transcript|biotype|strand|amino_acid_change|dna_change
The last three fields are omitted when empty. Consequences of compound variants which span multiple sites are printed in one record only, the remaining records link to it by '@position'. The consequence can start with the asterisk '*' prefix indicating a consequence downstream from a stop. For more details and examples please see the manual page and the split-vep plugin.
Performance
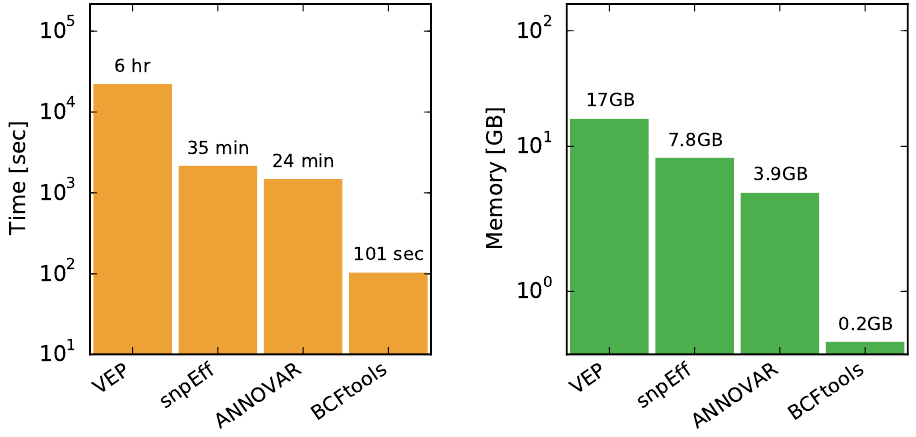
Performance comparison of BCFtools/csq with three popular consequence callers using a single-sample VCF with 4.5M sites. Note that the y-axis is logarithmic, BCFtools/csq requires fraction of CPU and memory required by other programs. The following program versions were used in the comparisoon: BCFtools 1.4, VEP v82, snpEff v4.2, ANNOVAR 2016Feb01.
References
Please cite this paper if you find our software useful doi: 10.1093/bioinformatics/btx100
Feedback
We welcome your feedback, please help us improve this page by either opening an issue on github or editing it directly and sending a pull request.